| Description: |
Canertinib is a pan-ErbB inhibitor for EGFR and ErbB2 with IC50 of 1.5 nM and 9.0 nM, no activity to PDGFR, FGFR, InsR, PKC, or CDK1/2/4. Canertinib displays anti-proliferative and pro-apoptotic effects in human myeloid leukemia cells devoid of ErbB-receptors, downregulates important signaling pathways and activates caspase-mediated intrinsic apoptosis pathway in human T-cell leukemia cells. |
| Targets: |
P-gp | Akt | ERK | STAT | PARP | Caspase | PI3K | MAPK | EGFR | ErbB2 |
| In vitro: |
| J. Med. Chem.,2013 May, 56(10):3820-32. | | Kinase scaffold repurposing for neglected disease drug discovery: discovery of an efficacious, lapatinib-derived lead compound for trypanosomiasis.[Pubmed: 23597080 ] | Human African trypanosomiasis (HAT) is a neglected tropical disease caused by the protozoan parasite Trypanosoma brucei . Because drugs in use against HAT are toxic and require intravenous dosing, new drugs are needed.
METHODS AND RESULTS:
Initiating lead discovery campaigns by using chemical scaffolds from drugs approved for other indications can speed up drug discovery for neglected diseases. We demonstrated recently that the 4-anilinoquinazolines lapatinib (GW572016, 1) and Canertinib (CI-1033) kill T. brucei with low micromolar EC50 values. We now report promising activity of analogues of 1, which provided an excellent starting point for optimization of the chemotype.
CONCLUSIONS:
Our compound optimization that has led to synthesis of several potent 4-anilinoquinazolines, including NEU617, 23a, a highly potent, orally bioavailable inhibitor of trypanosome replication. At the cellular level, 23a blocks duplication of the kinetoplast and arrests cytokinesis, making it a new chemical tool for studying regulation of the trypanosome cell cycle. | | Int J Pharm. 2012 Oct 15;436(1-2):127-34. | | Enhanced brain accumulation of pazopanib by modulating P-gp and Bcrp1 mediated efflux with canertinib or erlotinib.[Pubmed: 22688250] | Primary objective of this investigation was to delineate the differential impact of efflux transporters P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp1/Abcg2) on brain disposition and plasma pharmacokinetics of pazopanib. In addition, this research investigated whether inhibition of these efflux transporters with clinically relevant efflux modulators Canertinib or erlotinib could be a viable strategy for improving pazopanib brain delivery.
METHODS AND RESULTS:
In vitro assays with MDCKII cell monolayers suggested that pazopanib is a high affinity substrate for Bcrp1 and a moderate substrate for P-gp. Co-incubation with specific transport inhibitors restored cell accumulation and completely abolished the directionality of pazopanib flux. Brain and plasma pharmacokinetic studies were conducted in FVB wild type mice in the absence and presence of specific transport inhibitors. Drug levels in plasma and brain were determined using a validated high performance liquid chromatography method using vandetanib as an internal standard. In vivo studies indicated that specific inhibition of either P-gp (by zosuquidar or LY335979) or Bcrp1 (by Ko143) alone did not significantly alter pazopanib brain accumulation. However, dual P-gp/Bcrp1 inhibition by elacridar (GF120918), significantly enhanced pazopanib brain penetration by ~5-fold without altering its plasma concentrations. Thus, even though Bcrp1 showed higher affinity towards pazopanib in vitro, in vivo at the mouse BBB both P-gp and Bcrp1 act in concert to limit brain accumulation of pazopanib. Furthermore, erlotinib and Canertinib as clinically relevant efflux modulators efficiently abrogated directionality in pazopanib efflux in vitro and their co-administration resulted in 2-2.5-fold increase in pazopanib brain accumulation in vivo.
CONCLUSIONS:
Further pre-clinical and clinical investigations are warranted as erlotinib or Canertinib may have a synergistic pharmacological effect in addition to their primary role of pazopanib efflux modulation as a combination regimen for the treatment of recurrent brain tumors. |
|
| In vivo: |
| Br J Haematol. 2011 Oct;155(2):198-208. | | Irreversible pan-ERBB inhibitor canertinib elicits anti-leukaemic effects and induces the regression of FLT3-ITD transformed cells in mice.[Pubmed: 21848891] | Recent findings have indicated that tyrosine kinase inhibitors (TKIs) targeting the ERBB receptor family display anti-leukaemic effects, despite the lack of receptor expression on human leukaemic cells.
METHODS AND RESULTS:
The occurrence of activating mutations in the gene encoding FMS-like tyrosine kinase 3 (FLT3) in patients with acute myeloid leukaemia (AML) has rendered inhibition of this receptor a promising therapeutic target. Due to possibility of cross-reactivity, we investigated the effect of the irreversible pan-ERBB inhibitor Canertinib (CI-1033) on leukaemic cells expressing FLT3. The drug had anti-proliferative and apoptotic effects on primary AML cells and human leukaemic cell lines expressing mutated FLT3. In several AML patient samples, a blast cell population expressing FLT3-internal tandem duplication (ITD) was eradicated by Canertinib. Canertinib inhibited receptor autophosphorylation and kinase activity of both mutated and FLT3 ligand stimulated wildtype FLT3, leading to inhibition of the PI3-kinase and MAP kinase pathways. Apoptotic induction was dependent on pro-apoptotic BH3-only protein BCL2L11/BIM because siRNA silencing attenuated apoptosis. Moreover, the drug induced regression of cells expressing FLT3-ITD in a murine in vivo-transplantation model at previously described tolerated doses.
CONCLUSIONS:
These results indicate that Canertinib, as an irreversible TKI, could constitute a novel treatment regimen in patients with mutated or overexpressed FLT3. |
|




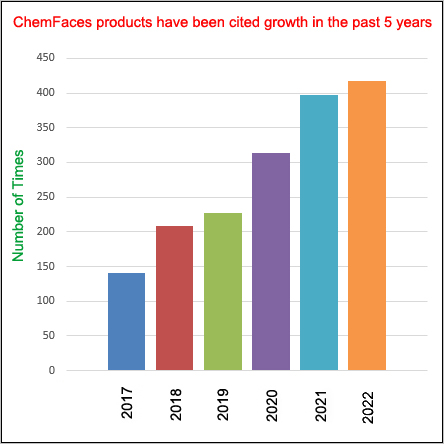
 Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019)
Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019) Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019)
Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019) Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)
Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)


 ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019)
ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019) Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019)
Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019) Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)
Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)