| Kinase Assay: |
| Drug Metab Dispos. 2006 Mar;34(3):449-56. | | Selectivity of substrate (trifluoperazine) and inhibitor (amitriptyline, androsterone, canrenoic acid, hecogenin, phenylbutazone, quinidine, quinine, and sulfinpyrazone) [Pubmed: 16381668] | Relatively few selective substrate and inhibitor probes have been identified for human UDP-glucuronosyltransferases (UGTs).
This work investigated the selectivity of trifluoperazine (TFP), as a substrate, and amitriptyline, androsterone, canrenoic acid, Hecogenin, phenylbutazone, quinidine, quinine, and sulfinpyrazone, as inhibitors, for human UGTs.
METHODS AND RESULTS:
Selectivity was assessed using UGTs 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7, and 2B15 expressed in HEK293 cells. TFP was confirmed as a highly selective substrate for UGT1A4. However, TFP bound extensively to both HEK293 lysate and human liver microsomes in a concentration-dependent manner (fuinc 0.20-0.59). When corrected for nonspecific binding, Km values for TFP glucuronidation were similar for both UGT1A4 (4.1 microM) and human liver microsomes (6.1+/-1.2 microM) as the enzyme sources. Of the compounds screened as inhibitors, Hecogenin, alone, was selective; significant inhibition was observed only for UGT1A4 (IC50 1.5 microM). Using phenylbutazone and quinine as "models," inhibition kinetics were variously described by competitive and noncompetitive mechanisms. Inhibition of UGT2B7 by quinidine was also investigated further, because the effects of this compound on morphine pharmacokinetics (a known UGT2B7 substrate) have been ascribed to inhibition of P-glycoprotein. Quinidine inhibited human liver microsomal and recombinant UGT2B7, with respective Ki values of 335+/-128 microM and 186 microM.
CONCLUSIONS:
In conclusion, TFP and Hecogenin represent selective substrate and inhibitor probes for UGT1A4, although the extensive nonselective binding of the former should be taken into account in kinetic studies. Amitriptyline, androsterone, canrenoic acid, Hecogenin, phenylbutazone, quinidine, quinine, and sulfinpyrazone are nonselective UGT inhibitors. |
|
| Cell Research: |
| Int J Oncol. 2003 Apr;22(4):899-905. | | Different contribution of apoptosis to the antiproliferative effects of diosgenin and other plant steroids, hecogenin and tigogenin, on human 1547 osteosarcoma cells.[Pubmed: 12632085] | Regulation of growth arrest and apoptosis are, in part, controlled by the tumor suppressor p53 after its phosphorylation which causes a determinant role in its functional activation. Moreover, PPAR regulate many functions such as proliferation and apoptosis.
METHODS AND RESULTS:
We compared the biological activity of diosgenin with Hecogenin and tigogenin, plant steroids structurally close to diosgenin, on proliferation rate, cell cycle distribution and apoptosis in human 1547 osteosarcoma cells. We found that all three molecules have an antiproliferative effect but gel shift analysis demonstrated that none of the plant steroids transactivated PPAR in human 1547 osteosarcoma cells whereas these molecules induced NF-kappaB binding to DNA. Although these plant steroids have a very close structure, only diosgenin caused a cell cycle arrest associated with strong apoptosis. This biological action seems correlated with a large increase of p53 protein expression.
CONCLUSIONS:
This fact was showed by immunofluorescence analysis which confirmed that diosgenin strongly enhanced the activation of p53 in contrast to Hecogenin and tigogenin actions. |
|
| Animal Research: |
| Eur J Pharmacol. 2012 May 15;683(1-3):260-9. | | Effects of hecogenin and its possible mechanism of action on experimental models of gastric ulcer in mice.[Pubmed: 22426163] | This study investigates the gastroprotective effects of Hecogenin, a steroid saponin isolated from Agave sisalana, on experimental models of gastric ulcer.
METHODS AND RESULTS:
Male Swiss mice were used in the models of ethanol- and indometacin-induced gastric ulcer.
To clarify the Hecogenin mechanism of action, the roles of nitric oxide (NO), sulfhydryls (GSH), K⁺(ATP) channels and prostaglandins were also investigated, and measurements of lipid peroxidation (TBARS assay) and nitrite levels in the stomach of Hecogenin-treated and untreated animals were performed. Furthermore, the effects of Hecogenin on myeloperoxidase (MPO) release from human neutrophils were assessed in vitro. Our results showed that Hecogenin (3.1, 7.5, 15, 30, 60 and 90 mg/kg, p.o.) acutely administered, before ethanol or indomethacin, exhibited a potent gastroprotective effect. Although the pretreatments with L-NAME, an iNOS inhibitor, and capsazepine, a TRPV1 receptor agonist, were not able to reverse the Hecogenin effect, this was reversed by glibenclamide, a K⁺(ATP) blocker, and indomethacin in the model of ethanol-induced gastric lesions. The Hecogenin pretreatment normalized GSH levels and significantly reduced lipid peroxidation and nitrite levels in the stomach, as evaluated by the ethanol-induced gastric lesion model. The drug alone increased COX-2 expression and this effect was further enhanced in the presence of ethanol. It also decreased MPO release and significantly protected the gastric mucosa.
CONCLUSIONS:
In conclusion, we showed that Hecogenin presents a significant gastroprotective effect that seems to be mediated by K⁺(ATP) channels opening and the COX-2/PG pathway. In addition, its antioxidant and anti-inflammatory properties may play a role in the gastroprotective drug effect. |
|




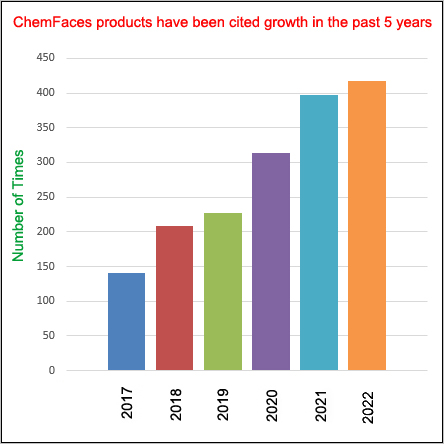
 Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019)
Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019) Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019)
Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019) Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)
Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)


 ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019)
ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019) Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019)
Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019) Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)
Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)