| In vitro: |
| Pharmaceutics . 2020 Sep 24;12(10):916. | | Quasi-Irreversible Inhibition of CYP2D6 by Berberine[Pubmed: 32987920] | | In our previous study, Hwang-Ryun-Hae-Dok-Tang, which contains berberine (BBR) as a main active ingredient, inhibited cytochrome P450 (CYP) 2D6 in a quasi-irreversible manner. However, no information is available on the detailed mechanism of BBR-induced CYP2D6 inhibition. Thus, the present study aimed to characterize the inhibition mode and kinetics of BBR and its analogues against CYP2D6 using pooled human liver microsomes (HLM). BBR exhibited selective quasi-irreversible inhibition of CYP2D6 with inactivation rate constant (kinact) of 0.025 min-1, inhibition constant (KI) of 4.29 μM, and kinact/KI of 5.83 mL/min/μmol. In pooled HLM, BBR was metabolized to Thalifendine (TFD), demethyleneberberine (DMB), M1 (proposed as demethylene-TFD), and to a lesser extent berberrubine (BRB), showing moderate metabolic stability with a half-life of 35.4 min and a microsomal intrinsic clearance of 7.82 μL/min/mg protein. However, unlike BBR, those metabolites (i.e., TFD, DMB, and BRB) were neither selective nor potent inhibitors of CYP2D6, based on comparison of half-maximal inhibitory concentration (IC50). Notably, TFD, but not DMB, exhibited metabolism-dependent CYP2D6 inhibition as in the case of BBR, which suggests that methylenedioxybenzene moiety of BBR may play a critical role in the quasi-irreversible inhibition. Moreover, the metabolic clearance of nebivolol (β-blocker; CYP2D6 substrate) was reduced in the presence of BBR. The present results warrant further evaluation of BBR-drug interactions in clinical situations. | | J Nat Prod . 2000 Dec;63(12):1638-40. | | In vitro antiplasmodial, antiamoebic, and cytotoxic activities of some monomeric isoquinoline alkaloids[Pubmed: 11141105] | | Twenty-one alkaloids have been assessed for activities against Plasmodium falciparum (multidrug- resistant strain K1) in vitro; 18 of these are reported for the first time. Two protoberberine alkaloids, dehydrodiscretine and berberine, were found to have antiplasmodial IC(50) values less than 1 M, while seven alkaloids-allocrytopine, columbamine, dehydroocoteine, jatrorrhizine, norcorydine, Thalifendine, and ushinsunine-had values between 1 and 10 M. These results are discussed in the context of structure-activity relationships. Compounds were also assessed for antiamoebic and cytotoxic activities, but none was significantly active except for berberine, which was moderately cytotoxic. | | J Transl Med . 2011 May 15;9:62. | | Bioactivities of berberine metabolites after transformation through CYP450 isoenzymes[Pubmed: 21569619] | | Background: Berberine (BBR) is a drug with multiple effects on cellular energy metabolism. The present study explored answers to the question of which CYP450 (Cytochrome P450) isoenzymes execute the phase-I transformation for BBR, and what are the bioactivities of its metabolites on energy pathways.
Methods: BBR metabolites were detected using LC-MS/MS. Computer-assistant docking technology as well as bioassays with recombinant CYP450s were employed to identify CYP450 isoenzymes responsible for BBR phase-I transformation. Bioactivities of BBR metabolites in liver cells were examined with real time RT-PCR and kinase phosphorylation assay.
Results: In rat experiments, 4 major metabolites of BBR, berberrubine (M1), Thalifendine (M2), demethyleneberberine (M3) and jatrorrhizine (M4) were identified in rat's livers using LC-MS/MS (liquid chromatography-tandem mass spectrometry). In the cell-free transformation reactions, M2 and M3 were detectable after incubating BBR with rCYP450s or human liver microsomes; however, M1 and M4 were below detective level. CYP2D6 and CYP1A2 played a major role in transforming BBR into M2; CYP2D6, CYP1A2 and CYP3A4 were for M3 production. The hepatocyte culture showed that BBR was active in enhancing the expression of insulin receptor (InsR) and low-density-lipoprotein receptor (LDLR) mRNA, as well as in activating AMP-activated protein kinase (AMPK). BBR's metabolites, M1-M4, remained to be active in up-regulating InsR expression with a potency reduced by 50-70%; LDLR mRNA was increased only by M1 or M2 (but not M3 and M4) with an activity level 35% or 26% of that of BBR, respectively. Similarly, AMPK-α phosphorylation was enhanced by M1 and M2 only, with a degree less than that of BBR.
Conclusions: Four major BBR metabolites (M1-M4) were identified after phase-I transformation in rat liver. Cell-free reactions showed that CYP2D6, CYP1A2 and CYP3A4 seemed to be the dominant CYP450 isoenzymes transforming BBR into its metabolites M2 and M3. BBR's metabolites remained to be active on BBR's targets (InsR, LDLR, and AMPK) but with reduced potency. |
|




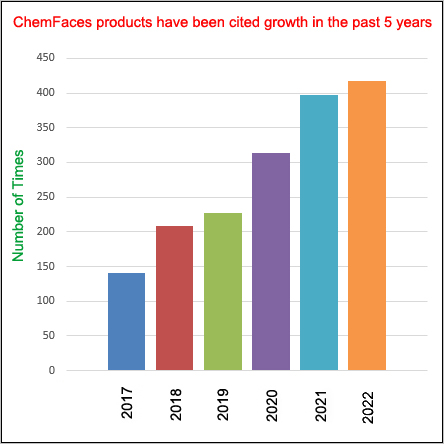
 Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019)
Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019) Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019)
Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019) Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)
Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)


 ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019)
ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019) Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019)
Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019) Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)
Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)