| In vitro: |
| J Virol. 2007 Feb;81(3):1186-1194. | | Rhinovirus activates interleukin-8 expression via a Src/p110beta phosphatidylinositol 3-kinase/Akt pathway in human airway epithelial cells[Pubmed: 17121804] | | Rhinovirus (RV) is responsible for the majority of common colds and triggers exacerbations of asthma and chronic obstructive lung disease. We have shown that RV serotype 39 (RV39) infection activates phosphatidylinositol 3 (PI 3)-kinase and the serine threonine kinase Akt minutes after infection and that the activation of PI 3-kinase and Akt is required for maximal interleukin-8 (IL-8) expression. Here, we further examine the contributions of Src and PI 3-kinase activation to RV-induced Akt activation and IL-8 expression. Confocal fluorescent microscopy of 16HBE14o- human bronchial epithelial cells showed rapid (10-min) colocalization of RV39 with Src, p85alpha PI 3-kinase, p110beta PI 3-kinase, Akt and Cit-Akt-PH, a fluorescent Akt pleckstrin homology domain which binds PI(3,4,5)P(3). The chemical Src inhibitor PP2 {4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo [3,4-d]pyrimidine} and the PI 3-kinase inhibitor LY294002 each inhibited Akt phosphorylation and the colocalization of RV39 with Akt. Digoxigenin-tagged RV coprecipitated with a Crosstide kinase likely to be Akt, and inhibition of Src blocked kinase activity. Digoxigenin-tagged RV39 colocalized with the lipid raft marker ceramide. In 16HBE14o- and primary mucociliary differentiated human bronchial epithelial cells, inhibition of Src kinase activity with the Src family chemical inhibitor PP2, dominant-negative Src (K297R), and Src small interfering RNA (siRNA) each inhibited RV39-induced IL-8 expression. siRNA against p110beta PI 3-kinase also inhibited IL-8 expression. These data demonstrate that, in the context of RV infection, Src and p110beta PI 3-kinase are upstream activators of Akt and the IL-8 promoter and that RV colocalizes with Src, PI 3-kinase, and Akt in lipid rafts. | | Biochim Biophys Acta. 2005 Oct 10;1725(3):340-347. | | Activation of a GST-tagged AKT2/PKBbeta[Pubmed: 15890450] | | The protein kinase AKT is a key regulator for cell growth, cell survival and metabolic insulin action. However, the mechanism of activation of AKT in vivo, which presumably involves membrane recruitment of the kinase, oligomerization, and multiple phosphorylation events, is not fully understood. In the present study, we have expressed and purified dimeric GST-fusion proteins of human protein kinase AKT2 (DeltaPH-AKT2) in milligram quantities via the baculovirus expression system. Treatment of virus-infected insect cells with the phosphatase inhibitor okadaic acid (OA) led to phosphorylation of the two regulatory phosphorylation sites, Thr309 and Ser474, and to activation of the kinase. Likewise, phosphorylation of Thr309 in vitro by recombinant PDK1 or mutation of Thr309 and Ser474 to acidic residues rendered the kinase constitutively active. However, even though the specific activity of our AKT2 was increased 15-fold compared to previous reports, GST-mediated dimerization alone did not lead to an activation of the kinase. Whereas both mutagenesis and phosphorylation led to an increase in the turnover number of the enzyme, only the latter resulted in a marked reduction (20-fold) of the apparent Km value for the exogenous substrate Crosstide, indicating that this widely used mutagenesis only partially mimics phosphorylation. Kinetic analysis of GST-AKT2 demonstrates that phosphorylation of Thr309 in the activation loop of the kinase is largely responsible for the observed reduction in Km and for a subsequent 150-fold increase in the catalytic efficiency (k(cat)/Km) of the enzyme. Highly active AKT2 constructs were used in autophosphorylation reactions in vitro, where inactive AKT2 kinases served as substrates. As a matter of fact, we found evidence for a minor autophosphorylation activity of AKT2 but no significant autophosphorylation of any of the two regulatory sites, Thr309 or Ser474. |
|




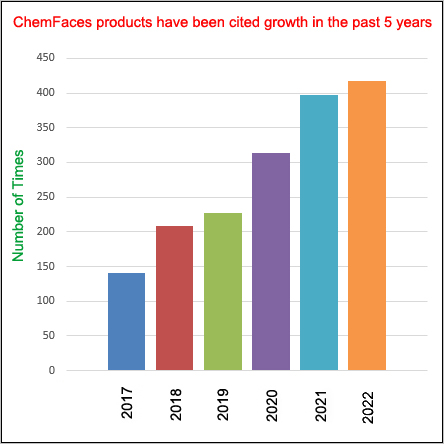
 Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019)
Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019) Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019)
Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019) Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)
Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)


 ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019)
ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019) Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019)
Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019) Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)
Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)