| In vitro: |
| Am J Clin Nutr. 2002 Nov;76(5):911-22. | | Fructose, weight gain, and the insulin resistance syndrome.[Pubmed: 12399260] | This review explores whether Fructose consumption might be a contributing factor to the development of obesity and the accompanying metabolic abnormalities observed in the insulin resistance syndrome.
METHODS AND RESULTS:
The per capita disappearance data for Fructose from the combined consumption of sucrose and high-Fructose corn syrup have increased by 26%, from 64 g/d in 1970 to 81 g/d in 1997. Both plasma insulin and leptin act in the central nervous system in the long-term regulation of energy homeostasis. Because Fructose does not stimulate insulin secretion from pancreatic beta cells, the consumption of foods and beverages containing Fructose produces smaller postprandial insulin excursions than does consumption of glucose-containing carbohydrate. Because leptin production is regulated by insulin responses to meals, Fructose consumption also reduces circulating leptin concentrations. The combined effects of lowered circulating leptin and insulin in individuals who consume diets that are high in dietary Fructose could therefore increase the likelihood of weight gain and its associated metabolic sequelae. In addition, Fructose, compared with glucose, is preferentially metabolized to lipid in the liver. Fructose consumption induces insulin resistance, impaired glucose tolerance, hyperinsulinemia, hypertriacylglycerolemia, and hypertension in animal models.
The data in humans are less clear.
CONCLUSIONS:
Although there are existing data on the metabolic and endocrine effects of dietary Fructose that suggest that increased consumption of Fructose may be detrimental in terms of body weight and adiposity and the metabolic indexes associated with the insulin resistance syndrome, much more research is needed to fully understand the metabolic effect of dietary Fructose in humans. |
|
| In vivo: |
| J Neurochem. 2015 May;133(4):572-81. | | Uptake and metabolism of fructose by rat neocortical cells in vivo and by isolated nerve terminals in vitro.[Pubmed: 25708447] | Fructose reacts spontaneously with proteins in the brain to form advanced glycation end products (AGE) that may elicit neuroinflammation and cause brain pathology, including Alzheimer's disease.
METHODS AND RESULTS:
We investigated whether Fructose is eliminated by oxidative metabolism in neocortex. Injection of [(14) C]Fructose or its AGE-prone metabolite [(14) C]glyceraldehyde into rat neocortex in vivo led to formation of (14) C-labeled alanine, glutamate, aspartate, GABA, and glutamine. In isolated neocortical nerve terminals, [(14) C]Fructose-labeled glutamate, GABA, and aspartate, indicating uptake of Fructose into nerve terminals and oxidative Fructose metabolism in these structures. This was supported by high expression of hexokinase 1, which channels Fructose into glycolysis, and whose activity was similar with Fructose or glucose as substrates. By contrast, the Fructose-specific ketohexokinase was weakly expressed. The Fructose transporter Glut5 was expressed at only 4% of the level of neuronal glucose transporter Glut3, suggesting transport across plasma membranes of brain cells as the limiting factor in removal of extracellular Fructose. The genes encoding aldose reductase and sorbitol dehydrogenase, enzymes of the polyol pathway that forms glucose from Fructose, were expressed in rat neocortex. These results point to Fructose being transported into neocortical cells, including nerve terminals, and that it is metabolized and thereby detoxified primarily through hexokinase activity. We asked how the brain handles Fructose, which may react spontaneously with proteins to form 'advanced glycation end products' and trigger inflammation. Neocortical cells took up and metabolized extracellular Fructose oxidatively in vivo, and isolated nerve terminals did so in vitro.
CONCLUSIONS:
The low expression of Fructose transporter Glut5 limited uptake of extracellular Fructose.
Hexokinase was a main pathway for Fructose metabolism, but ketohexokinase (which leads to glyceraldehyde formation) was expressed too. Neocortical cells also took up and metabolized glyceraldehyde oxidatively. | | J Clin Endocrinol Metab. 2004 Jun;89(6):2963-72. | | Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women.[Pubmed: 15181085 ] | Previous studies indicate that leptin secretion is regulated by insulin-mediated glucose metabolism. Because Fructose, unlike glucose, does not stimulate insulin secretion, we hypothesized that meals high in Fructose would result in lower leptin concentrations than meals containing the same amount of glucose.
METHODS AND RESULTS:
Blood samples were collected every 30-60 min for 24 h from 12 normal-weight women on 2 randomized days during which the subjects consumed three meals containing 55, 30, and 15% of total kilocalories as carbohydrate, fat, and protein, respectively, with 30% of kilocalories as either a Fructose-sweetened [high Fructose (HFr)] or glucose-sweetened [high glucose (HGl)] beverage. Meals were isocaloric in the two treatments. Postprandial glycemic excursions were reduced by 66 +/- 12%, and insulin responses were 65 +/- 5% lower (both P < 0.001) during HFr consumption. The area under the curve for leptin during the first 12 h (-33 +/- 7%; P < 0.005), the entire 24 h (-21 +/- 8%; P < 0.02), and the diurnal amplitude (peak - nadir) (24 +/- 6%; P < 0.0025) were reduced on the HFr day compared with the HGl day. In addition, circulating levels of the orexigenic gastroenteric hormone, ghrelin, were suppressed by approximately 30% 1-2 h after ingestion of each HGl meal (P < 0.01), but postprandial suppression of ghrelin was significantly less pronounced after HFr meals (P < 0.05 vs. HGl). Consumption of HFr meals produced a rapid and prolonged elevation of plasma triglycerides compared with the HGl day (P < 0.005).
CONCLUSIONS:
Because insulin and leptin, and possibly ghrelin, function as key signals to the central nervous system in the long-term regulation of energy balance, decreases of circulating insulin and leptin and increased ghrelin concentrations, as demonstrated in this study, could lead to increased caloric intake and ultimately contribute to weight gain and obesity during chronic consumption of diets high in Fructose. | | J. Clin. Invest., 2009, 119(5):1322-34. | | Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans.[Pubmed: 19381015 ] | Studies in animals have documented that, compared with glucose, dietary Fructose induces dyslipidemia and insulin resistance.
METHODS AND RESULTS:
To assess the relative effects of these dietary sugars during sustained consumption in humans, overweight and obese subjects consumed glucose- or Fructose-sweetened beverages providing 25% of energy requirements for 10 weeks. Although both groups exhibited similar weight gain during the intervention, visceral adipose volume was significantly increased only in subjects consuming Fructose. Fasting plasma triglyceride concentrations increased by approximately 10% during 10 weeks of glucose consumption but not after Fructose consumption. In contrast, hepatic de novo lipogenesis (DNL) and the 23-hour postprandial triglyceride AUC were increased specifically during Fructose consumption. Similarly, markers of altered lipid metabolism and lipoprotein remodeling, including fasting apoB, LDL, small dense LDL, oxidized LDL, and postprandial concentrations of remnant-like particle-triglyceride and -cholesterol significantly increased during Fructose but not glucose consumption. In addition, fasting plasma glucose and insulin levels increased and insulin sensitivity decreased in subjects consuming Fructose but not in those consuming glucose.
CONCLUSIONS:
These data suggest that dietary Fructose specifically increases DNL, promotes dyslipidemia, decreases insulin sensitivity, and increases visceral adiposity in overweight/obese adults. |
|




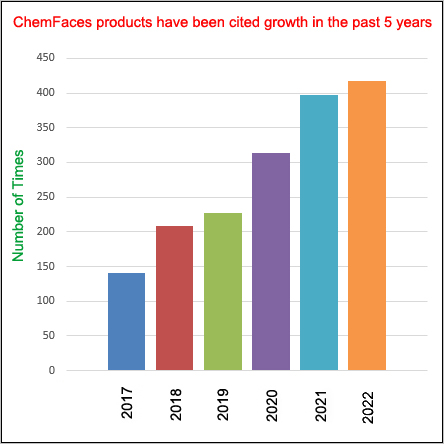
 Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019)
Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019) Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019)
Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019) Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)
Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)


 ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019)
ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019) Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019)
Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019) Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)
Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)