| In vitro: |
| Toxicol Lett. 2015 May 5;234(3):162-71. | | GAPDH-knockdown reduce rotenone-induced H9C2 cells death via autophagy and anti-oxidative stress pathway.[Pubmed: 25725130] | GAPDH, well known for its house-keeping functions, has also been shown to be involved in cell injury, apoptosis and death under conditions of stress such as starvation, chemical injury and oxidative stress. This study examines the effect of GAPDH knockdown on cell injury in response to Rotenone.
METHODS AND RESULTS:
GAPDH was knocked down in H9C2 cardiomyoblasts using siRNA prior to exposure to Rotenone (0 nM, 20 nM, 40 nM and 80 nM). Autophagy was detected by western blot for autophagy proteins (Beclin-1, Atg5, LC-3A/B and p62) and MDC staining for acidic substances. Pro-apoptosis protein and flow cytometry were used to assess cell apoptosis and death and intracellular ATP relative concentration was measured. Oxidant stress was assessed by measuring DCFH-DA, TBARS, GSH and SOD.
In this study, GAPDH-knockdown enhanced autophagy in Rotenone-induced H9C2 cells, decreased oxidant stress and increased antioxidant pathways; and reduced cell apoptosis and death. Furthermore, GAPDH-knockdown preserved cell energy.
CONCLUSIONS:
siRNA-mediated GAPDH knockdown reduced Rotenone-induced H9C2 cell death occurring via autophagy and anti-oxidative stress pathway. This study enriches the understanding of GAPDH pathophysiology role, and provides potential new therapeutic targets for cardiac disease states characterized by oxidative stress. | | Toxicology. 2015 Feb 3;328:75-81. | | JNK inhibition of VMAT2 contributes to rotenone-induced oxidative stress and dopamine neuron death.[Pubmed: 25496994] | Treatment with Rotenone, both in vitro and in vivo, is widely used to model dopamine neuron death in Parkinson's disease upon exposure to environmental neurotoxicants and pesticides. Mechanisms underlying Rotenone neurotoxicity are still being defined.
METHODS AND RESULTS:
Our recent studies suggest that Rotenone-induced dopamine neuron death involves microtubule destabilization, which leads to accumulation of cytosolic dopamine and consequently reactive oxygen species (ROS). Furthermore, the c-Jun N-terminal protein kinase (JNK) is required for Rotenone-induced dopamine neuron death. Here we report that the neural specific JNK3 isoform of the JNKs, but not JNK1 or JNK2, is responsible for this neuron death in primary cultured dopamine neurons. Treatment with taxol, a microtubule stabilizing agent, attenuates Rotenone-induced phosphorylation and presumably activation of JNK. This suggests that JNK is activated by microtubule destabilization upon Rotenone exposure. Moreover, Rotenone inhibits VMAT2 activity but not VMAT2 protein levels. Significantly, treatment with SP600125, a pharmacological inhibitor of JNKs, attenuates Rotenone inhibition of VMAT2. Furthermore, decreased VMAT2 activity following in vitro incubation of recombinant JNK3 protein with purified mesencephalic synaptic vesicles suggests that JNK3 can inhibit VMAT2 activity.
CONCLUSIONS:
Together with our previous findings, these results suggest that Rotenone induces dopamine neuron death through a series of sequential events including microtubule destabilization, JNK3 activation, VMAT2 inhibition, accumulation of cytosolic dopamine, and generation of ROS. Our data identify JNK3 as a novel regulator of VMAT2 activity. |
|




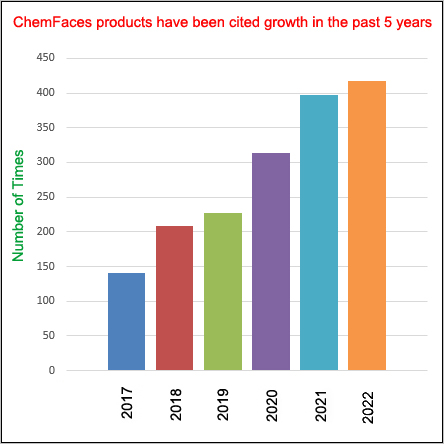
 Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019)
Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019) Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019)
Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019) Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)
Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)


 ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019)
ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019) Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019)
Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019) Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)
Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)