| In vitro: |
| PLoS One. 2013 Dec 9;8(12):e81220. | | Norisoboldine suppresses VEGF-induced endothelial cell migration via the cAMP-PKA-NF-κB/Notch1 pathway.[Pubmed: 24349042 ] | The migration of endothelial cells has been regarded as a potential target for the treatment of angiogenesis-related diseases. Previously, we demonstrated that Norisoboldine (NOR), an alkaloid compound isolated from Radix Linderae, can significantly suppress synovial angiogenesis by selectively inhibiting endothelial cell migration.
METHODS AND RESULTS:
In this study, we evaluated the importance of various pathways in VEGF-induced endothelial cell migration using specific inhibitor. VEGF-induced endothelial cell migration and sprouting were significantly inhibited by H-89 (an inhibitor of protein kinase A (PKA)) but not by inhibitors of other pathways. NOR markedly suppressed VEGF-induced intracytoplasmic cAMP production and PKA activation and thereby down-regulated the activation of downstream components of the PKA pathway, including enzymes (src, VASP and eNOS) and the transcription factor NF-κB. Moreover, the transcription activation potential of NF-κB, which is related to IκBα phosphorylation and the disruption of the p65/IκBα complex, was reduced by NOR. Meanwhile, NOR selectively inhibited the expression of p-p65 (ser276) but not p-p65 (ser536) or PKAc, indicating that PKAc participates in the regulation of NF-κB by NOR. Co-immunoprecipitation and immunofluorescence assays confirmed that NOR inhibited the formation of the PKAc/p65 complex and thereby decreased p65 (ser276) phosphorylation to prevent p65 binding to DNA. Docking models indicated that the affinity of NOR for PKA was higher than that of the original PKA ligand. Moreover, the fact that H-89 improved Notch1 activation, but DAPT (an inhibitor of Notch) failed to affect PKA activation, suggested that PKA may act on upstream of Notch1. In conclusion, the inhibitory effects of NOR on endothelial cell migration can be attributed to its modulation of the PKA pathway, especially on the processes of p65/IκBα complex disruption and PKAc/p65 complex formation.
CONCLUSIONS:
These results suggest that NOR inhibit VEGF-induced endothelial cell migration via a cAMP-PKA-NF-κB/Notch1 signaling pathway. |
|
| In vivo: |
| Int Immunopharmacol. 2014 May;20(1):110-6. | | Norisoboldine induces apoptosis of fibroblast-like synoviocytes from adjuvant-induced arthritis rats.[Pubmed: 24613208] | Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by pronounced synovial inflammation and hyperplasia, in which there may be an imbalance between the growth and death of fibroblast-like synoviocytes (FLS). Norisoboldine (NOR), the main active constituent in the alkaloid fraction isolated from Radix Linderae, was previously demonstrated to alleviate arthritis severity in experimental RA.
METHODS AND RESULTS:
This study aimed to evaluate the effects of NOR on proliferation and apoptosis of FLS from adjuvant-induced arthritis (AIA) rats to elucidate the mechanism of its inhibitory effect on inflammatory synovial hyperplasia in RA. Our results indicated that NOR exhibited a pro-apoptotic effect on AIA FLS but only slightly affected cell proliferation and the cell cycle. Following treatment with NOR for 24h, the activation of caspase 3 and caspase 9 and the cleavage of poly (ADP-ribose) polymerase (PARP) in AIA FLS were observed; however, caspase 8 remained unaffected. Meanwhile, a flow cytometric assay revealed that NOR significantly increased the percentage of apoptotic cells, causing the loss of the depolarized mitochondrial membrane potential and the release of cytochrome C. The expression of Bax and Bcl-2 was also regulated by NOR treatment. Additionally, the expression of p53 protein was up-regulated by NOR, and pretreatment with PFT-α, a p53 specific inhibitor, reversed the increase in FLS apoptosis caused by NOR.
CONCLUSIONS:
These findings indicated that NOR-induced apoptosis in AIA FLS is achieved via a mitochondrial-dependent pathway, which may be mediated by promoting the release of cytochrome C and by regulating the expression of Bax and Bcl-2 proteins, and p53 might also be required for NOR-induced apoptosis in AIA FLS. | | Eur J Pain. 2014 Aug;18(7):939-48. | | Norisoboldine attenuates inflammatory pain via the adenosine A1 receptor.[Pubmed: 24395183] | Norisoboldine (NOR) is a benzylisoquinoline alkaloid isolated from Radix Linderae, a traditional Chinese medicine. Our previous studies have demonstrated that it produces anti-inflammatory and anti-rheumatoid arthritis effects.
METHODS AND RESULTS:
The present study was undertaken to explore the analgesic effects of NOR and its potential mechanism in the formalin test and the acetic acid writhing test.
Oral administration of NOR dose dependently attenuated the formalin-induced pain responses in the second phase, and reduced formalin-induced paw oedema. It also diminished acetic acid-induced writhing responses but had no effect on acute thermal pain in the hotplate test. The mechanistic studies suggested that the adenosine system, but not the opioid receptor system, is involved in NOR-induced antinociception. Naloxone, a non-selective opioid receptor antagonist, had no effect on NOR-induced analgesic action. However, caffeine (a non-selective adenosine receptor antagonist) completely reversed the analgesic effect of NOR in formalin-induced nociceptive responses in the second phase, and 8-cyclopentyl-1, 3-dipropylxanthine (DPCPX, a selective adenosine A1 receptor antagonist) completely inhibited NOR-induced analgesia in both formalin-induced nociceptive responses and acetic acid-induced writhing responses. In addition, NOR reduced formalin-induced activation of extracellular signal-regulated kinase and calcium/calmodulin-dependent protein kinase II in the spinal cord, which is also blocked by DPCPX. Furthermore, NOR decreased forskolin-evoked cyclic adenosine monophosphate levels in mouse spinal cord neuronal cultures through the adenosine A1 receptor.
CONCLUSIONS:
Our data demonstrate that NOR produces the analgesic effect in inflammatory pain by a mechanism related to the adenosine system. | | Cell Death Dis . 2018 Feb 15;9(3):258. | | Norisoboldine, a natural AhR agonist, promotes Treg differentiation and attenuates colitis via targeting glycolysis and subsequent NAD +/SIRT1/SUV39H1/H3K9me3 signaling pathway[Pubmed: 29449535] | | Abstract
Norisoboldine (NOR), a natural aryl hydrocarbon receptor (AhR) agonist, has been demonstrated to attenuate ulcerative colitis (UC) and induce the generation of Treg cells. Under UC condition, hypoxia widely exists in colonic mucosa, and secondary changes of microRNAs (miRs) expressions and glycolysis contribute to Treg differentiation. At present, we worked for exploring the deep mechanisms for NOR-promoted Treg differentiation in hypoxia and its subsequent anti-UC action from the angle of AhR/miR or AhR/glycolysis axis. Results showed that NOR promoted Treg differentiation in hypoxia and the effect was stronger relative to normoxia. It activated AhR in CD4+ T cells under hypoxic microenvironment; CH223191 (a specific AhR antagonist) and siAhR-3 abolished NOR-promoted Treg differentiation. Furthermore, the progress of glycolysis, levels of Glut1 and HK2, and expression of miR-31 rather than miR-219 and miR-490 in CD4+ T cells were downregulated by NOR treatment under hypoxic microenvironment. However, HK2 plasmid but not miR-31 mimic significantly interfered NOR-enhanced Treg polarization. In addition, NOR reduced NAD+ and SIRT1 levels, facilitated the ubiquitin-proteasomal degradation of SUV39H1 protein, and inhibited the enrichment of H3K9me3 at -1, 201 to -1,500 region of Foxp3 promoter in CD4+ T cells under hypoxic microenvironment, which was weakened by HK2 plasmid, CH223191, and siAhR-3. Finally, the correlation between NOR-mediated activation of AhR, repression of glycolysis, regulation of NAD+/SIRT1/SUV39H1/H3K9me3 signals, induction of Treg cells, and remission of colitis was confirmed in mice with DSS-induced colitis by using CH223191 and HK2 plasmid. In conclusion, NOR promoted Treg differentiation and then alleviated the development of colitis by regulating AhR/glycolysis axis and subsequent NAD+/SIRT1/SUV39H1/H3K9me3 signaling pathway. |
|




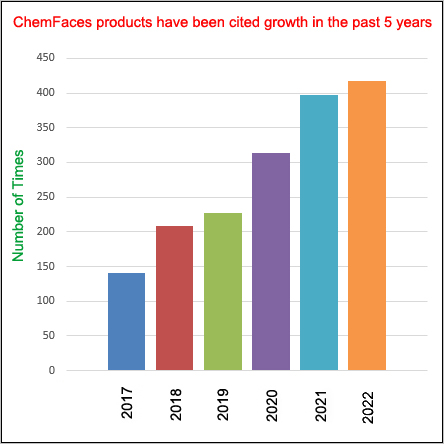
 Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019)
Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019) Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019)
Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019) Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)
Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)


 ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019)
ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019) Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019)
Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019) Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)
Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)