| Kinase Assay: |
| Fitoterapia. 2013 Mar;85:109-13. | | Cryptotanshinone and dihydrotanshinone I exhibit strong inhibition towards human liver microsome (HLM)-catalyzed propofol glucuronidation.[Pubmed: 23333907] | Danshen is one of the most famous herbs in the world, and more and more danshen-prescribed drugs interactions have been reported in recent years. Evaluation of inhibition potential of danshen's major ingredients towards UDP-glucuronosyltransferases (UGTs) will be helpful for understanding detailed mechanisms for danshen-drugs interaction. Therefore, the aim of the present study is to investigate the inhibitory situation of cryptotanshinone and Dihydrotanshinone I towards UGT enzyme-catalyzed propofol glucuronidation.
METHODS AND RESULTS:
In vitro the human liver microsome (HLM) incubation system was used, and the results showed that cryptotanshinone and Dihydrotanshinone I exhibited dose-dependent inhibition towards HLM-catalyzed propofol glucuronidation. Dixon plot and Lineweaver-Burk plot showed that the inhibition type was best fit to competitive inhibition type for both cryptotanshinone and Dihydrotanshinone I. The second plot using the slopes from the Lineweaver-Burk plot versus the concentrations of cryptotanshinone or Dihydrotanshinone I was employed to calculate the inhibition parameters (Ki) to be 0.4 and 1.7μM, respectively. Using the reported maximum plasma concentration (Cmax), the altered in vivo exposure of propofol increased by 10% and 8.2% for the co-administration of Dihydrotanshinone I and cryptotanshinone, respectively.
CONCLUSIONS:
All these results indicated the possible danshen-propofol interaction due to the inhibition of Dihydrotanshinone I and cryptotanshinone towards the glucuronidation reaction of propofol. | | J Mol Neurosci. 2014 Jul;53(3):506-10. | | Acetylcholinesterase complexes with the natural product inhibitors dihydrotanshinone I and territrem B: binding site assignment from inhibitor competition and validation through crystal structure determination.[Pubmed: 24573600] | Acetylcholinesterase (AChE) is a critical enzyme that regulates neurotransmission by degrading the neurotransmitter acetylcholine in synapses of the nervous system. It is an important target for both therapeutic drugs that treat Alzheimer's disease and organophosphate (OP) chemical warfare agents that cripple the nervous system and cause death through paralysis.
METHODS AND RESULTS:
We are exploring a strategy to design compounds that bind tightly at or near a peripheral or P-site near the mouth of the AChE active site gorge and exclude OPs from the active site while interfering minimally with the passage of acetylcholine. However, to target the AChE P-site, much more information must be gathered about the structure-activity relationships of ligands that bind specifically to the P-site. Here, we review our recent reports on two uncharged, natural product inhibitors of AChE, Dihydrotanshinone I and territrem B, that have relatively high affinities for the enzyme. We describe an inhibitor competition assay and comment on the structures of these inhibitors in complex with recombinant human acetylcholinesterase as determined by X-ray crystallography.
CONCLUSIONS:
Our results reveal that Dihydrotanshinone I binding is specific to only the P-site, while territrem B binding spans the P-site and extends into the acylation or A-site at the base of the gorge. | | Int Immunopharmacol. 2015 Sep;28(1):764-72. | | Blockade of TNF-α-induced NF-κB signaling pathway and anti-cancer therapeutic response of dihydrotanshinone I.[Pubmed: 26283590] | The nuclear factor-κB (NF-κB) transcription factors control many physiological processes including inflammation, immunity, apoptosis, and angiogenesis. We identified Dihydrotanshinone I as an inhibitor of NF-κB activation through our research on Salvia miltiorrhiza Bunge.
METHODS AND RESULTS:
In this study, we found that Dihydrotanshinone I significantly inhibited the expression of NF-κB reporter gene induced by TNF-α in a dose-dependent manner. And Dihydrotanshinone I also inhibited TNF-α induced phosphorylation and degradation of IκBα, phosphorylation and nuclear translocation of p65. Furthermore, pretreatment of cells with this compound prevented the TNF-α-induced expression of NF-κB target genes, such as anti-apoptosis (cIAP-1 and FLIP), proliferation (COX-2), invasion (MMP-9), angiogenesis (VEGF), and major inflammatory cytokines (TNF-α, IL-6, and MCP1). We also demonstrated that Dihydrotanshinone I potentiated TNF-α-induced apoptosis. Moreover, Dihydrotanshinone I significantly impaired activation of extracellular signal-regulated kinase 1/2 (ERK1/2), p38 and stress-activated protein kinase/c-Jun NH2-terminal kinase (JNK/SAPK). In vivo studies demonstrated that Dihydrotanshinone I suppressed the growth of HeLa cells in a xenograft tumor model, which could be correlated with its modulation of TNF-α production.
CONCLUSIONS:
Taken together, Dihydrotanshinone I could be a valuable candidate for the intervention of NF-κB-dependent pathological conditions such as inflammation and cancer. | | Latin Am. J. Pharm., 2012, 31(7):1060-3. | | Dihydrotanshinone I Exhibits Strong Inhibition Towards UDP-glucuronosyltransferase (UGT) 1A7.[Reference: WebLink] | Inhibition of the activity of UDP-glucuronosyltransferases (UGTs) can induce severe drugdrug interaction and metabolic disorders of endogenous substances. The aim of the present study is to investigate the inhibition of important UGT isoforms by Dihydrotanshinone I, which is an important bioactive component isolated from danshen.
METHODS AND RESULTS:
The nonselective probe substrate 4-methylumbelliferone (4-MU), and the recombinant UGT isoforms were used in the present study. The results showed that 100 M of Dihydrotanshinone I inhibited the activity of UGT1A1, UGT1A3, UGT1A6, UGT1A7, UGT1A8, UGT1A10, and UGT2B7 by 32.7, 61.5, 61.1, 77.5, 47.9, 62.8, and 55.9 %, respectively. Further inhibition kinetic study was performed for the inhibition of UGT1A7 by Dihydrotanshinone I. Dose-dependent inhibition of UGT1A7 by Dihydrotanshinone I was detected, and Dixon and Lineweaver-Burk plots showed that the inhibition of UGT1A7 by Dihydrotanshinone I was best fit to competitive inhibition type. The inhibition kinetic parameter (Ki) was determined to be 2.8 μM. Using the in vivo maximum plasma concentration (Cmax) of Dihydrotanshinone I (11.29 ng/mL, 0.04 μM), the the change of AUC ranged from 0.14 to 1.42 % when the contribution of UGT1A7 towards the metabolism of drugs (fm) ranged from 0.1 to 1.
CONCLUSIONS:
Given that UGT1A7 is one of the most important gastrointestinal UGT isoforms and has high correlation with the occurence of cancer, the potential danshen-drug interaction due to the inhibition of UGT1A7 by Dihydrotanshinone I should be given more attention. |
|
| Cell Research: |
| J Biosci Bioeng. 2000;89(3):292-3. | | Biological activity of dihydrotanshinone I: effect on apoptosis.[Pubmed: 16232748] | Recently, we have found for the first time that Dihydrotanshinone I, isolated from Salvia miltiorrhiza, exhibited cytotoxicity against various tumor cell lines.
METHODS AND RESULTS:
To investigate whether the mechanism underlying Dihydrotanshinone I toxicity involved apoptosis in cancer cell lines, we examined cell growth arrest and cell death by flow cytometric analysis and DNA fragmentation assay. Dihydrotanshinone I induced cell growth arrest during the S phase and subsequently, apoptosis, following its application to K562/ADR cells, whereas cryptotanshinone did not have these effects.
CONCLUSIONS:
These results suggest that the mode of action of Dihydrotanshinone I involves apoptotic pathways that are different from those involved in cryptotanshinone toxicity. |
|




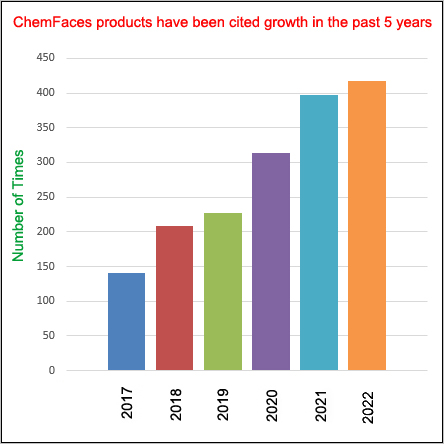
 Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019)
Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019) Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019)
Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019) Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)
Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)


 ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019)
ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019) Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019)
Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019) Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)
Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)