| Kinase Assay: |
| Biochemical Pharmacology, 18 Dec 2015, 102:45-63. | | Cucurbitacin I elicits the formation of actin/phospho-myosin II co-aggregates by stimulation of the RhoA/ROCK pathway and inhibition of LIM-kinase.[Reference: WebLink] | Cucurbitacins are cytotoxic triterpenoid sterols isolated from plants. One of their earliest cellular effect is the aggregation of actin associated with blockage of cell migration and division that eventually lead to apoptosis.
METHODS AND RESULTS:
We unravel here that Cucurbitacin I actually induces the co-aggregation of actin with phospho-myosin II. This co-aggregation most probably results from the stimulation of the Rho/ROCK pathway and the direct inhibition of the LIMKinase. We further provide data that suggest that the formation of these co-aggregates is independent of a putative pro-oxidant status of Cucurbitacin I.
CONCLUSIONS:
The results help to understand the impact of cucurbitacins on signal transduction and actin dynamics and open novel perspectives to use it as drug candidates for cancer research. | | Cancer Biology & Therapy, 01 Jan 2015, 16(2):233-243. | | Cucurbitacin-I inhibits Aurora kinase A, Aurora kinase B and survivin, induces defects in cell cycle progression and promotes ABT-737-induced cell death in a caspase-independent manner in malignant human glioma cells.[Reference: WebLink] | Because STAT signaling is commonly activated in malignant gliomas as a result of constitutive EGFR activation, strategies for inhibiting the EGFR/JAK/STAT cascade are of significant interest.
METHODS AND RESULTS:
We, therefore, treated a panel of established glioma cell lines, including EGFR overexpressors, and primary cultures derived from patients diagnosed with glioblastoma with the JAK/STAT inhibitor Cucurbitacin I. Treatment with cucurbitacin-I depleted p-STAT3, p-STAT5, p-JAK1 and p-JAK2 levels, inhibited cell proliferation, and induced G2/M accumulation, DNA endoreduplication, and multipolar mitotic spindles. Longer exposure to Cucurbitacin I significantly reduced the number of viable cells and this decrease in viability was associated with cell death, as confirmed by an increase in the subG1 fraction.
CONCLUSIONS:
Our data also demonstrated that cucurbitacin-I strikingly downregulated Aurora kinase A, Aurora kinase B and survivin. We then searched for agents that exhibited a synergistic effect on cell death in combination with cucurbitacin-I. We found that cotreatment with cucurbitacin-I significantly increased Bcl(-)2/Bcl(-)xL family member antagonist ABT-737-induced cell death regardless of EGFR/PTEN/p53 status of malignant human glioma cell lines. Although >50% of the cucurbitacin-I plus ABT-737 treated cells were annexin V and propidium iodide positive, PARP cleavage or caspase activation was not observed. Pretreatment of z-VAD-fmk, a pan caspase inhibitor did not inhibit cell death, suggesting a caspase-independent mechanism of cell death. Genetic inhibition of Aurora kinase A or Aurora kinase B or survivin by RNA interference also sensitized glioma cells to ABT-737, suggesting a link between STAT activation and Aurora kinases in malignant gliomas. |
|




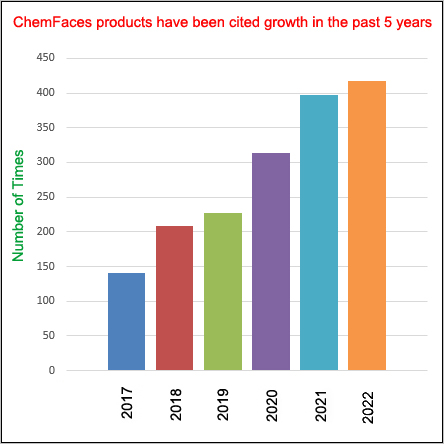
 Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019)
Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019) Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019)
Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019) Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)
Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)


 ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019)
ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019) Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019)
Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019) Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)
Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)